Sequenciamento no AM revela linhagem diferente de coronavírus achado em SP
Sem tempo, irmão
- Fiocruz Amazônia encontra 11 mutações do novo coronavírus; 9 diferem dos da China
- Isso não significa necessariamente vírus mais letais, diz coordenador da pesquisa
- Para ele, pesquisas em diferentes estados do Brasil ajudam a entender epidemia
- Vírus pode mudar de acordo com fatores como clima, mas ainda faltam mais estudos
O primeiro sequenciamento do coronavírus feito na Amazônia revelou que ele já tem 11 mutações em relação ao que foi sequenciado em São Paulo, em fevereiro. Isso aponta para a circulação de linhagens diferentes do vírus no país.
A diferença encontrada entre os vírus dentro do nosso país é maior do que as do vírus original de Wuhan, na China, onde foram registrados os primeiros casos da covid-19. Ao todo, foram nove mutações diferentes achadas no coronavírus da Amazônia em comparação ao chinês.


A pesquisa foi feita na Fiocruz (Fundação Oswaldo Cruz) Amazônia, em Manaus, e coordenada pelo cientista na área de virologia e biologia molecular Felipe Naveca. A amostra do vírus foi feita no dia 16 de março com material colhido de um paciente infectado na Espanha.
"A gente não tem ainda como saber se essas mutações da China para cá já são alguma coisa que terá impacto do ponto de vista clínico", conta.
O sequenciamento deixa claro que o vírus está em evolução —o que não quer dizer necessariamente que esteja causando problemas mais graves no ser humano. "A priori não tem nada que a gente possa dizer que a mutação aqui está relacionada a um aumento de virulência desse vírus", diz Naveca. "Mas isso sugere que ele ainda deve evoluir muito", conta o cientista.

Leia a entrevista completa:
Tilt - Pelo que o senhor analisou, esse vírus passou por mutações até chegar aí na Amazônia?
Felipe Naveca - Nós fizemos o genoma completo dessa amostra que chegou aqui no Amazonas no dia 15 de março. No dia 16, nós coletamos amostra, e alguns dias depois a gente já tinha o genoma. Esse paciente ele veio da Espanha, e essa amostra tinha nove mutações em relação ao genoma original da amostra de Wuhan, na China.
Tilt - O que essas nove mudanças querem dizer? Mudaram para serem mais "fortes"?
Felipe Naveca - Os vírus, como os outros organismos, passam sempre por processo evolutivo. O que acontece nos vírus RNA, como é o coronavírus e o dengue também, é que essa evolução acontece mais rápido por causa da própria característica biológica de produzir o RNA. Essa enzima não tem correção. Então, como ela não corrige, cada vez que é produzido um RNA pode juntar um erro —o que seriam as mutações— e esses erros se perpetuam da seguinte maneira: se for um que seja um problema para o vírus, ele vai sumir porque aquilo não vai funcionar. Se essa mutação for benéfica ou neutra, ela pode ser perpetuada.
A gente não tem ainda como saber exatamente se essas mutações da China para cá já são alguma coisa que terá impacto do ponto de vista clínico. O que precisa ser feito agora é juntar o maior banco de dados possível —e é o que está sendo feito no mundo inteiro— para a gente poder avaliar e ter um suporte estatístico. Quer dizer, se encontrarmos uma mutação mais vezes em casos mais graves, a gente pode, no futuro, associar essa mutação a um desfecho pior.
Tilt - O senhor viu alguma mudança ao que foi achado em São Paulo?
Felipe Naveca - Em relação à amostra sequenciada pelo pessoal do Albert Einstein, que foi publicado no dia 2 de março, de um paciente coletado no dia 28 de fevereiro, tem 11 mutações.
É importante essa análise do sequenciamento não só para a gente saber exatamente o caminho que o vírus fez; ela também nos serve para montar esse banco de dados para, futuramente, no dia que nós tivermos uma droga, ou no dia que nós tivermos uma vacina, saber se existem o que a gente chama de escape, que é um vírus que é resistente ao tratamento ou escapa de uma vacina.
Isso a gente só vai saber na hora que juntarmos os dados que estão sendo feitos mundialmente. É um trabalho que nunca foi feito nessa velocidade. Há três meses que a gente tem o conhecimento desse vírus e já temos mais de 1.000 genomas do mundo inteiro disponíveis. E a gente poderá ter um banco de dado bastante robusto para, quando a gente tiver uma droga ou uma vacina, saber identificar aqueles que possam eventualmente escapar.
Tilt - Dá para dizer que essa diferença de 11 em relação a São Paulo é normal ou surpreende?
Felipe Naveca - Esperado para este tipo de vírus, mas sugere que ele ainda deve evoluir muito.

Tilt - Mas já dá para saber se teremos diferença do vírus que faz a transmissão comunitária por aí na Amazônia, por exemplo, levando em conta calor, umidade, presença de florestas?
Felipe Naveca - O vírus vai evoluir de acordo com aquela população por onde ele vai ser transmitido. Então a gente imagina que vão ter diferenças. A gente chama isso de linhagem, que vai ser mais comum nessas amostras que circulam aqui na Amazônia. Na verdade, é mais provável que a gente tenha mais de uma introdução do vírus no mesmo estado. Provavelmente uma delas vai ser mais sucesso em se estabelecer na população.
Você vai ter uma variação aqui que não é exatamente a mesma variação em São Paulo, então em um país grande e com população miscigenada como o nosso é importante você ter essa amostragem de sequências não só de um lugar. É por isso que a gente está fazendo aqui também na região Norte.
Quanto à questão do clima, a gente sabe que nas áreas ou épocas mais frias há uma tendência a ter maior número de casos de vírus respiratórios, seja do coronavírus ou dos outros que a gente já conhecia. Nós temos agora essa época de chuvas, no chamado inverno amazônico, onde também há a circulação desses vírus. O clima sozinho não vai ser o suficiente para conter esta circulação.
Tilt - O que chamou a atenção do senhor, como virologista, do vírus sequenciado na Amazônia?
Felipe Naveca - Como é um vírus ainda muito novo, a gente ainda não tem muito com o que comparar para dizer que é diferente, ou que chama atenção em comparação a um outro vírus. O que a gente está comparando são as sequências desse vírus do próprio coronavírus do final de 2019. A priori não tem nada que a gente possa dizer que a mutação aqui está relacionada a um aumento de virulência desse vírus.
Tilt - Mas o que 11 diferenças em um mesmo país quer dizer? Temos, por exemplo, mais de um tipo de vírus no país?
Felipe Naveca - Não dá para falar em dois tipos, como a gente fala, por exemplo, os [vírus da Aids] HIV 1, HIV 2. Mas a gente pode dizer, sim, que tem diferenças de linhagens nas amostras que entraram no Brasil em diferentes regiões. Certamente teremos amostras que entraram nos Estados Unidos, que passaram por um processo evolutivo, que entraram diretamente da China. Ou seja, temos a mesma situação de pessoas que vieram da Itália, da Espanha, de outros lugares.
Provavelmente a gente vai descobrir daqui a algumas semanas, quando aumentar esse número de dados sequenciados, é que nós tivemos diversas introduções do mesmo vírus, mas com pequenas variações entre elas, que a gente pode chamar de linhagens.
Tilt - O Brasil recebeu esse vírus depois de passar por muitos países. Ter mais linhagens é bom ou ruim?
Felipe Naveca - O fato de nós termos sido atingidos posteriormente só dá uma expectativa de entender melhor o quão grave essa doença poderia ser. Nos deu mais tempo de preparação. Acho que nesse ponto, sim, é bom. E também a gente tem essa diferença de que atingiu a Europa mais no inverno deles, e nós ainda não estamos nessa fase.
A gente espera passar do pico antes de chegarmos, por exemplo, no auge do nosso inverno. Vamos ver como é que isso vai seguir. Sobre as linhagens, pode ser que uma delas consiga prosperar melhor no país. Mas a gente ainda não tem informação suficiente para falar isso.
Tilt - Então seria certo dizer que, por ter mais linhagens vindas de vários locais, teremos mais "riscos" dele se estabelecer aqui por meio de uma seleção natural?
Felipe Naveca - Podemos dizer que recebemos diferentes linhagens do vírus —assim como os EUA, que também está mais ou menos na mesma época [do grau de epidemia] e aumentando o número de casos— que já evoluíram nesses três meses. Certamente esse vírus já é diferente do primeiro [encontrado pela ciência] porque ali ainda não era um que estava em seres humanos. Mas sim, a gente está recebendo várias linhagens que já passaram por esse processo de coevolução.
Mas nem sempre a evolução em um sistema hospedeiro —no caso, nós, humanos— vai tornar ele mais agressivo. Por exemplo, se você for olhar como exemplo os hantavírus que evoluíram em roedores, naquelas espécies ele não causa problema. Nem sempre um vírus que está evoluindo no ser humano vai ser mais agressivo a ele.
Tilt - Pelo que o senhor está vendo, esse vírus está se adaptando bem em todos os climas do Brasil, não é?
Felipe Naveca - Sim, não parece que o clima seja uma barreira muito importante, senão não teríamos tantos casos.
Tilt - Aí no Norte, região quente e úmida, é propícia para vírus respiratórios normalmente?
Felipe Naveca - Nós temos tido muitos casos nos últimos anos, incluindo casos graves por vírus sincicial respiratório e Influenza, Influenza H1N1 e Influenza B.

Tilt - Existe um risco de termos vírus se proliferando juntos?
Felipe Naveca - Sim, certamente está ocorrendo. Algumas pessoas podem se infectar com mais de um vírus ao mesmo tempo.
Tilt - O que acontece nesses casos?
Felipe Naveca - Nós temos uma tendência a imaginar que seria um quadro pior, mas o fato é que temos que investigar em mais casos se há essa coinfecção. Com poucos casos descritos não dá para generalizar se sempre é mais grave.
Tilt - Qual é a necessidade de seguir com as pesquisas do coronavírus pelo país?
Felipe Naveca - Estudos desse tipo devem ser realizados em diferentes regiões, pois certamente temos diferentes introduções do vírus, que possivelmente vão evoluir de forma diferente, nas diferentes populações atingidas. Além disso, é importante destacar que esses estudos no Brasil só foram possíveis em tão pouco tempo porque a comunidade científica estava preparada para responder. E em ciência isso significa infraestrutura, capacitação e insumos, ou seja, investimento em pesquisa. Aqui, por exemplo, já havíamos montado uma rede genômica de pesquisa em saúde, para responder a emergências como esta.
SIGA TILT NAS REDES SOCIAIS
- Twitter: https://twitter.com/tilt_uol
- Instagram: https://www.instagram.com/tilt_uol/
- WhatsApp: https://uol.page.link/V1gDd
- Grupo no Facebook Deu Tilt: http://bit.ly/FacebookTilt







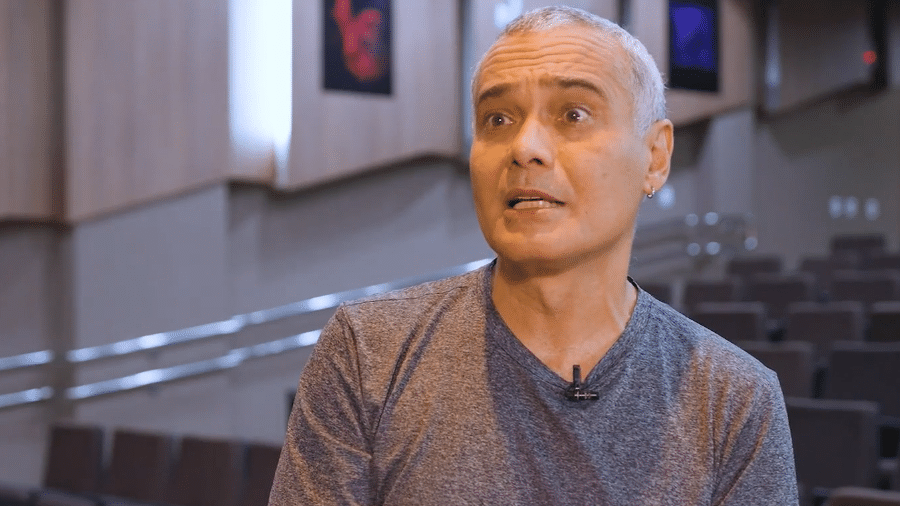







ID: {{comments.info.id}}
URL: {{comments.info.url}}
Ocorreu um erro ao carregar os comentários.
Por favor, tente novamente mais tarde.
{{comments.total}} Comentário
{{comments.total}} Comentários
Seja o primeiro a comentar
Essa discussão está encerrada
Não é possivel enviar novos comentários.
Essa área é exclusiva para você, assinante, ler e comentar.
Só assinantes do UOL podem comentar
Ainda não é assinante? Assine já.
Se você já é assinante do UOL, faça seu login.
O autor da mensagem, e não o UOL, é o responsável pelo comentário. Reserve um tempo para ler as Regras de Uso para comentários.