Saiba o que é exigido para a aprovação de vacinas no Brasil

Nos últimos meses, só se fala em vacina pra lá e pra cá. A pandemia do novo coronavírus acelerou os processos de produção dos produtos e fez com que a população se interessasse pelo tema. Produzir um novo imunizante não é tarefa simples, exige pesquisa por anos a fio e muitos passos até chegar, com segurança, ao braço de todo mundo.
Como todo medicamento, elas são importantes para o controle de doenças. Antes de chegar à população, porém, precisam passar por estudos realizados pelas empresas que desenvolvem o produto para a comprovação de sua qualidade, segurança e eficácia.


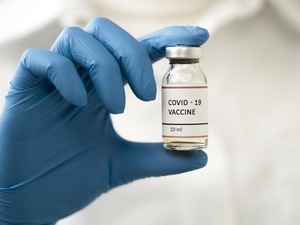
No Brasil, a Anvisa (Agência Nacional de Vigilância Sanitária) é o órgão responsável pela avaliação e aprovação de solicitações para a realização de pesquisas clínicas com fins de registro e de pedidos de registro de imunobiológicos desenvolvidos pela indústria farmacêutica.
No entanto, para que a agência possa fazer a análise do produto, é necessário que as empresas demonstrem interesse, registrando a solicitação de avaliação de estudo clínico ou de registro junto ao órgão.
Saiba abaixo quais são as etapas desse processo, desde os estudos laboratoriais, passando pelos testes em animais e humanos, até a chegada do pedido de registro à Anvisa.
Primeira etapa
Durante o desenvolvimento do produto, um dos primeiros passos da trajetória até o registro é a realização de estudos da fase não clínica, aplicada em animais de experimentação, de acordo com protocolos estabelecidos pela empresa.
Esta etapa, que ocorre em laboratórios, precede testes em humanos e tem como objetivo investigar a ação e a segurança da molécula em laboratório, procedimento comum exigido para qualquer tipo de novo medicamento.
Nesta fase, a vacina passa por ensaios que auxiliam os pesquisadores a verificar a dose adequada a ser administrada e a conhecer o mecanismo de ação do produto, bem como determinar sua segurança e imunogenicidade, antes de passar aos testes em humanos.
Fases dos estudos em humanos
Depois de cumprir as etapas da fase não clínica, a empresa deve realizar estudos clínicos em três fases (1, 2 e 3) para avaliar e determinar a segurança e a eficácia do uso da vacina em humanos. Cada etapa cumpre objetivos específicos no âmbito do desenvolvimento do produto.
Durante a fase 1, pequenos grupos de indivíduos, normalmente adultos saudáveis, são avaliados para verificação da segurança e determinação do tipo de resposta imune provocada pela vacina. Nessa fase também podem ser realizados estudos de desafio, a fim de selecionar os melhores projetos para seguirem à fase seguinte.
Na fase 2, há a inclusão de um maior número de indivíduos e o produto já é administrado a indivíduos representativos da população-alvo (bebês, crianças, adolescentes, adultos, idosos ou imunocomprometidos). Nessa fase é avaliada a segurança da vacina, a imunogenicidade, a posologia e o modo de administração.
Na fase 3, a vacina é administrada a uma grande quantidade de indivíduos, normalmente milhares de pessoas, para que seja demonstrada a sua eficácia e segurança, ou seja, que a vacina é capaz de proteger os indivíduos com o mínimo possível de reações adversas.
O início dos testes em seres humanos depende, além das aprovações ética e regulatória, da própria organização interna dos pesquisadores para recrutamento dos voluntários.
Autorização para estudos em humanos
Para realização de qualquer pesquisa clínica envolvendo seres humanos, é obrigatória a aprovação dos Comitês de Ética em Pesquisa (CEPs) e/ou da Comissão Nacional de Ética em Pesquisa (Conep).
A anuência de pesquisa pela Anvisa se aplica somente aos estudos clínicos que têm a finalidade de registro e pós-registro de medicamentos, incluindo vacinas, sempre por solicitação da empresa patrocinadora ou de seu representante. É importante esclarecer que o prazo para início da pesquisa, após a aprovação ética e regulatória, é definido pelo patrocinador do estudo.
Dossiê de Desenvolvimento Clínico de Medicamentos
A realização de estudos clínicos de fase 1, 2 e 3, para fins de registro e pós-registro, deve ser aprovada antes pela Anvisa. Para isso, a empresa desenvolvedora da pesquisa deve submeter à agência o DDCM (Dossiê de Desenvolvimento Clínico de Medicamentos), que traz informações detalhadas sobre o medicamento e sobre o estudo.
Para dar maior celeridade à avaliação de solicitações para a realização de pesquisas no Brasil, neste momento de pandemia de covid-19, a Anvisa instituiu o Comitê de Avaliação de Estudos Clínicos, Registro e Pós-Registro de Medicamentos para prevenção ou tratamento da Covid-19, o que inclui também as vacinas.
O comitê é responsável por analisar os pedidos de anuência de estudos clínicos de produtos para prevenção e tratamento da Covid-19, de forma prioritária, no prazo médio de até 72 horas após a submissão formal do protocolo, entre outras atribuições.
Registro de vacinas
Em primeiro lugar, as empresas do setor farmacêutico devem manifestar interesse em fazer o registro. Somente a partir do momento em que há essa manifestação, por meio do peticionamento do pedido junto à Anvisa, é que o processo de regularização tem início.
Segundo a norma, além de apresentar documentos, plano de farmacovigilância e informações gerais sobre o produto, a empresa deve entregar relatório com dados sobre as matérias-primas utilizadas na vacina, como a descrição das cepas, sua origem, identificação, processos de obtenção ou construção, entre diversos outros dados.
É importante frisar que a empresa pode solicitar avaliação de registro de produtos com estudos da fase 3 em andamento, portanto já com os dados consolidados das fases 1 e 2, desde que seja demonstrada uma alta eficácia terapêutica ou preventiva e/ou não exista outra terapia ou droga alternativa comparável para aquele estágio da doença.
Outro item importante é que, além da pesquisa clínica, a desenvolvedora da vacina também realiza, paralelamente, o estudo de estabilidade do medicamento para gerar dados sobre, por exemplo, o prazo de validade e as condições de armazenamento adequadas.
Portanto, a solicitação de registro de uma vacina só é aceita se a empresa apresentar toda a documentação exigida. Além disso, quem desenvolve o produto deve garantir pesquisas pós-comercialização, como forma de monitoramento, por exemplo, de novas reações adversas não detectadas nos estudos anteriores.
Prazos e priorização da análise
De acordo com a Anvisa, o prazo para a primeira manifestação sobre o pedido de registro de vacinas enquadradas é de até 60 dias. Neste período, a agência pode conceder o registro, solicitar informações complementares para análise ou, ainda, indeferir o pedido —este prazo refere-se exclusivamente ao registro.
Quando se trata de solicitação para a realização de estudos clínicos para covid-19, o prazo médio para a manifestação da Anvisa é de até 72 horas.
*Com informações da Anvisa.











ID: {{comments.info.id}}
URL: {{comments.info.url}}
Ocorreu um erro ao carregar os comentários.
Por favor, tente novamente mais tarde.
{{comments.total}} Comentário
{{comments.total}} Comentários
Seja o primeiro a comentar
Essa discussão está encerrada
Não é possivel enviar novos comentários.
Essa área é exclusiva para você, assinante, ler e comentar.
Só assinantes do UOL podem comentar
Ainda não é assinante? Assine já.
Se você já é assinante do UOL, faça seu login.
O autor da mensagem, e não o UOL, é o responsável pelo comentário. Reserve um tempo para ler as Regras de Uso para comentários.