Padre tem doença que atinge 1 a cada 1 milhão de pessoas: "Sou milionário"
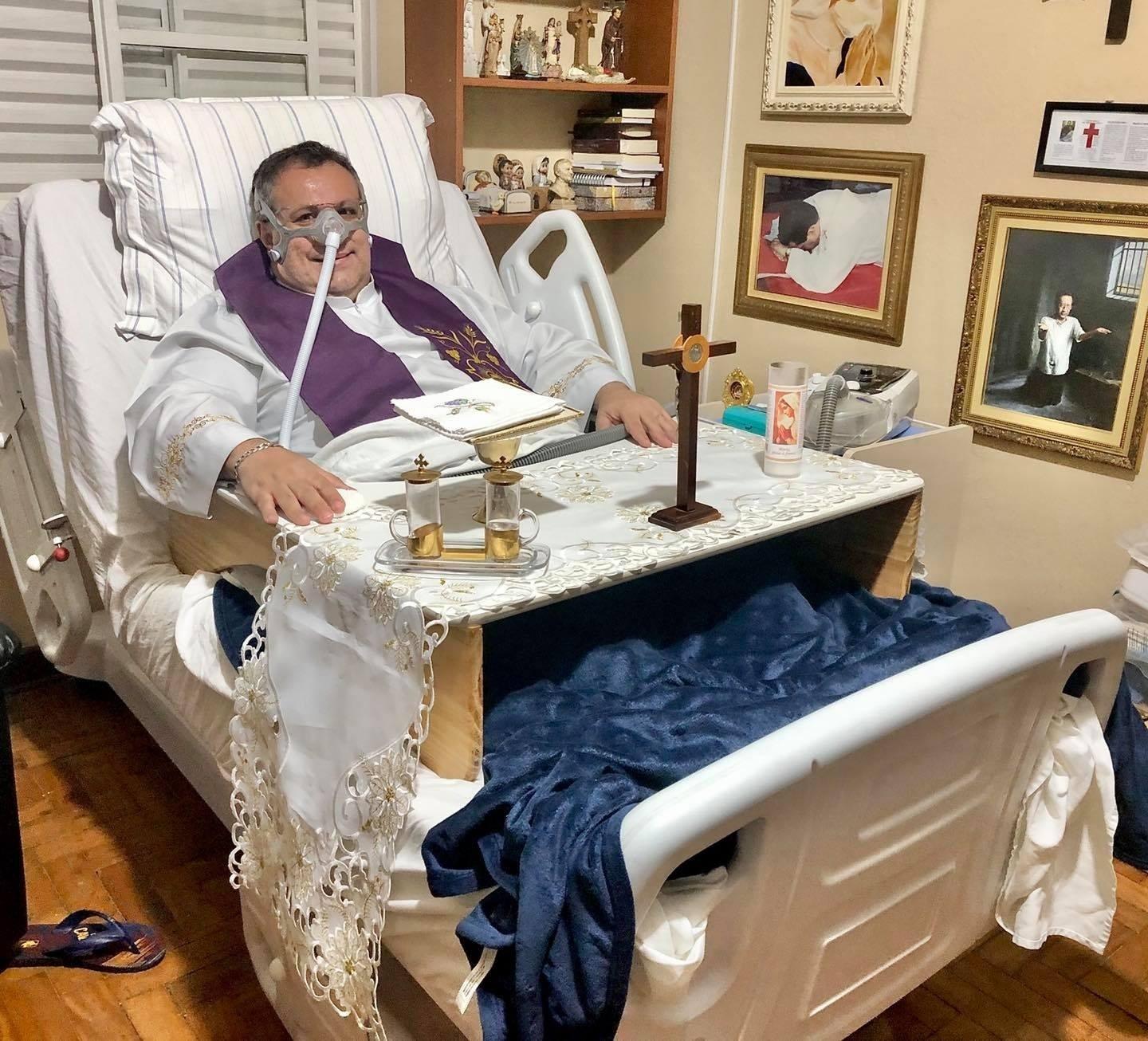











Com uma forma branda de uma doença rara, mas que vem avançando nos últimos tempos, o padre Márlon Múcio, 47, teve vários sintomas ao longo da vida, mas só em 2019, após nove anos de investigação, ele descobriu que tem a Deficiência do Transportador de Riboflavina (RTD). Segundo estimativas da Cure RTD Foundation, nos Estados Unidos, são 300 casos diagnosticados no mundo e nove no Brasil. A cada um milhão de pessoas, apenas uma tem RTD. Ao levar a doença com fé e bom humor, o padre Márlon brinca que é um milionário.
"Sem saber, tive os primeiros sintomas da RTD ainda criança. Aos sete anos, perdi a audição e precisei fazer uma cirurgia corretiva. Aos 14, sentia muito cansaço ao mastigar. Apesar desses problemas de saúde levava a minha vida normalmente.



Tinha uma rotina ativa, celebrava a missa na Casa João Paulo 2º, em Taubaté, em São Paulo, onde moro, viajava por vários países pregando retiros, participava de programas de rádio e TV, escrevi e publiquei 35 livros cristãos, com três milhões de exemplares vendidos.

A partir de 2010, alguns sintomas se intensificaram e outros começaram a aparecer. O primeiro deles foram as quedas frequentes na rua, teve um dia que quase fui atropelado.
Outro sintoma que inclusive me assustou bastante era não conseguir mais segurar a cabeça, ficava com o pescoço como o de um recém-nascido.
A dificuldade para mastigar e engolir piorou e passei a engasgar muito. Uma fraqueza generalizada se instalou nos braços e nas pernas. Tenho muita fadiga, só de falar ou me vestir, por exemplo, já fico exaustivamente cansado. Recentemente, minha parte motora ficou tão comprometida a ponto de eu andar cambaleando e precisar usar o andador e a cadeira de rodas.
Passar por todas essas coisas foi impactante, tive que me adaptar a várias situações, desde usar um respirador 24 horas por dia por causa do cansaço e da falta de ar até não conseguir mais andar.
Um dos momentos mais difíceis para mim foi celebrar a missa, pela primeira vez, de andador. Fiquei com vergonha, me senti fracassado, frustrado, incapaz e exposto. Não conseguia olhar para os fiéis.
Lembro que cheguei em casa e a minha sobrinha, Mariana, que na época tinha uns oito anos, me disse: 'Tio padre, não fica tristinho, você explicou o evangelho tão direitinho, todo mundo entendeu. Você é o meu tio padre favorito de todas as galáxias, o mais lindo'. Eu sorri e, na missa seguinte, voltei para a igreja com o andador e de cabeça erguida.
Nenhum médico sabia o que eu tinha
De 2010 a 2019, iniciei uma grande investigação em busca de um diagnóstico. Fiz vários exames e passei em diversos médicos, entre clínico geral, ortopedista, neurologista, psiquiatra, otorrino, geneticista... Às vezes passava com mais de um médico da mesma especialidade, mas ninguém descobria qual doença eu tinha. Isso causava uma grande angústia e sofrimento em mim e na minha família.

Nunca me entreguei à enfermidade, mas chegou um momento em que fiquei cansado de tudo e não queria ir a mais nenhuma consulta. Recebi o diagnóstico errado de cinco doenças: miastenia gravis, miastenia congênita, canalopatia, doença de pompe e doença mitocondrial.
Fiz os tratamentos, mas como não respondia às medicações na comparação com os outros pacientes, os médicos descartaram as doenças.
Estava em uma corrida contra o tempo, minha saúde se deteriorando cada vez mais. Como ninguém sabia o que eu tinha, houve um momento em que imaginei que as pessoas poderiam pensar que eu era doido ou queria chamar a atenção.
Desde 2014, meu quadro piorou e fui internado várias vezes. Em uma dessas internações, em 2016, tive uma experiência de quase céu. Alguns médicos podem pensar que foi o excesso de remédios ou algum tipo de confusão mental, mas sei que vi os meus santos de devoção pegando a minha maca e me levando para o céu.
Vi os portões dourados do céu, mas não entrei. Os santos me disseram que teria que voltar, que ainda não era a minha hora. Acredito que fui barrado porque minha missão na terra ainda não acabou, muitas pessoas oraram por mim e ainda preciso santificar ainda mais e fazer mais boas obras.
Depois de muita insistência, passei com um segundo geneticista em junho de 2018. Fui em várias consultas, fiz um monte de exames e, quase um ano depois, em março de 2019, recebi o diagnóstico de Deficiência do Transportador de Riboflavina (RTD) após nove anos de buscas. Foi um alívio, finalmente faria o tratamento correto.
Apesar de todas as limitações e das dores horríveis que sinto no tórax, na cintura, nos membros, no rosto e de viver à base de morfina, sou grato a Deus por essa doença não afetar a minha parte cognitiva. Levo a RTD com fé, leveza, alegria e bom humor.
Às vezes, choro e pareço que estou bravo nas fotos, com cara de buldogue por causa dos músculos caídos no rosto e da fraqueza facial, mas é só impressão. Brinco que sou um milionário, a cada um milhão de pessoas, apenas uma tem a minha doença.
Faço o tratamento medicamentoso, além de fisioterapia motora e respiratória todos os dias, e fonoterapia três vezes por semana, além de acompanhamento com diversos médicos. Meus pais e meu irmão são meus cuidadores e conto com um serviço de homecare.

Celebrava missa para multidões, mas por causa da fraqueza, desde abril deste ano tenho celebrado da cama, só para os meus familiares, mas faço com o mesmo amor e dedicação, pois sei que posso alcançar o mundo pelo efeito espiritual da missa.
Além disso, levo uma palavra de esperança sobre fé e superação da doença através das redes sociais. Quando jovem, meu sonho era ser médico. Após uma comunhão na missa, ouvi de Jesus que eu seria médico sim, mas de almas —o que de fato me tornei.
Eu e os outros pacientes criamos a página Cure RTD Brasil no Facebook e no Instagram (@curertdbrasil), com o objetivo dar apoio às pessoas com RTD e seus familiares, auxiliar no diagnóstico, conscientizar os profissionais de saúde e a população em geral, bem como arrecadar fundos para pesquisas que levem a um tratamento mais efetivo.
A Cure RTD Brasil está ligada a Cure RTD Foundation, nos Estados Unidos, e ao Instituto Vidas Raras. Já sou um milagre vivo, não tenho medo de morrer, estou em profunda paz com Deus. Minha missão é ajudar a descobrir a cura para essa doença".
Saiba mais sobre a RTD
1) O que é a Deficiência do Transportador de Riboflavina?
É uma doença rara, de origem genética, causada por alteração em um gene do DNA. Esses pacientes têm a falta de uma enzima que transporta a riboflavina para dentro da célula que está funcionando de forma inadequada. Por conta disso, a pessoa com RTD tem falta de riboflavina intracelular e não consegue manter todas as atividades metabólicas adequadas.
A pessoa com RTD nasce com a doença, ela não pode ser adquirida como se fosse uma doença infecciosa. Apesar de ser genética, não quer dizer que é necessário ter história na família, mesmo os pais, geralmente, não são afetados por ela. Isso acontece porque ela é uma doença de herança recessiva, isto é, o paciente com RTD precisa ter herdado tanto da mãe como do pai a alteração no gene do transportador da riboflavina. Apesar de ter uma cópia do gene com a alteração, os pais costumam ter uma outra cópia normal, dessa forma compensando a cópia alterada e, por isso, eles não desenvolvem a doença. O maior risco em relação à recorrência de RTD é para os irmãos de um paciente já diagnosticado.
2) Quais os tipos de RTD?
Existem dois tipos principais de RTD: a Síndrome de Brown-Vialetto-Van Laere e a Síndrome de Fazio-Londe. Ambos são causadas por defeito no transporte da riboflavina e têm sintomas semelhantes. A principal diferença é que a Síndrome de Fazio-Londe não tem acometimento de um nervo craniano muito importante. Na Síndrome de Brown-Vialetto-Van Laere, esse nervo está sempre acometido, o que faz com que as pessoas desenvolvam perda auditiva.
3) Quais os sintomas da RTD?
Os principais sintomas são fraqueza muscular em mãos, pés, braços, pernas, pescoço e tronco. Uma marcha atáxica, ou seja, dificuldade de andar com perda do equilíbrio também é um fator comum. É possível notar uma movimentação involuntária dos olhos, o que chamamos de nistagmo, e uma movimentação da língua semelhante a um tremor, que chamamos de fasciculação. Sintomas como dificuldade de deglutir alimentos sólidos e líquidos são frequentes. Por fim, a fraqueza da musculatura responsável pela respiração pode levar à insuficiência respiratória.
O início desses sintomas pode ser variável. Algumas pessoas podem ser assintomáticas até a adolescência ou vida adulta. Outras podem manifestar sintomas já ao nascimento ou nos primeiros dias de vida, bem como na infância, como uma pequena perda auditiva, uma leve fraqueza ou cansaço, e só apresentar a evolução da doença na vida adulta.
Outros sintomas iniciais incluem fraqueza da musculatura facial, o que gera um semblante muitas vezes "cansado", "triste" ou "desanimado", e dificuldades na fala, com uma fala um pouco mais arrastada e dificuldades de completar palavras. Algumas alterações de capacidade visual também podem estar presentes.
4) Quais as maiores limitações que a doença impõe ao paciente?
Quando não controlada com medicação e apoio terapêutico, a evolução da doença pode levar a incapacidade de andar. Os sintomas respiratórios, devido à fraqueza da musculatura respiratória, podem levar a parada respiratória e ser potencialmente fatal. O uso de suporte ventilatório, como alguns tipos de respiradores, tende a trazer um conforto para o paciente, já que dá suporte para o pulmão, diminuindo a força que os músculos precisam fazer para o pulmão se encher de ar.
5) Como é feito diagnóstico?
O diagnóstico, inicialmente, é feito por uma suspeita clínica com base na história pessoal e familiar do indivíduo, além dos sintomas. Exames de sangue específicos para avaliar algumas substâncias do metabolismo são realizados, no entanto, é o exame genético que confirma a RTD: o sequenciamento dos genes do transporte da riboflavina, SLC52A2 e SLC52A3. O exame é uma coleta de sangue, ou de saliva, do qual, no laboratório, é extraído o DNA e sequenciado esses dois genes em busca de variantes que confirmem o diagnóstico.
Na ausência do exame genético, outros podem auxiliar no diagnóstico, como a eletromiografia, que avalia os sinais musculares, e o eletroencefalograma, que avalia a atividade do cérebro. Exames para avaliação auditiva e oftalmológica também devem ser considerados. A polissonografia, que verifica a qualidade do sono e se ocorrem apneias (pequenas "paradas" respiratórias durante a noite), também é importante.
6) Quais os tratamentos?
O principal tratamento é suplementação de riboflavina, em altas doses, e pela vida toda. As terapias de suporte e reabilitação são essenciais, incluindo fonoterapia, fisioterapia, terapia ocupacional e psicoterapia.
7) A RTD tem cura?
A RTD não tem cura, porém com o tratamento adequado e correto, em conjunto com uma equipe multidisciplinar, é possível ter o controle da doença, evitando sua progressão. Pessoas com quadros mais leves podem levar uma vida normal. Em casos mais graves ou debilitantes, a qualidade de vida pode ser prejudicada, e a adaptação de atividades e ritmos de vida são fundamentais para os pacientes.
Fonte: Rodrigo Ambrosio Fock, médico geneticista do Centro de Genética Médica da Unifesp (Universidade Federal de São Paulo), mestre em tecnologias e atenção à saúde e membro titular da Sociedade Brasileira de Genética Médica e Genômica.











ID: {{comments.info.id}}
URL: {{comments.info.url}}
Ocorreu um erro ao carregar os comentários.
Por favor, tente novamente mais tarde.
{{comments.total}} Comentário
{{comments.total}} Comentários
Seja o primeiro a comentar
Essa discussão está encerrada
Não é possivel enviar novos comentários.
Essa área é exclusiva para você, assinante, ler e comentar.
Só assinantes do UOL podem comentar
Ainda não é assinante? Assine já.
Se você já é assinante do UOL, faça seu login.
O autor da mensagem, e não o UOL, é o responsável pelo comentário. Reserve um tempo para ler as Regras de Uso para comentários.