Com 1 ano, ela foi diagnosticada com doença ultrarrara e sem tratamento





Cerca de seis meses após nascer, a pequena Isadora Bender Prateleira, hoje com 9 anos, começou a ter vômito e diarreia constantes e, logo na sequência, episódios de infecção respiratória. Depois de meses de idas e vindas aos hospitais e consultórios médicos, ela recebeu o diagnóstico de ASMD (acid sphingomyelinase deficiency ou deficiência de esfingomielinase ácida, em português), uma doença ultrarrara que até setembro de 2022 não tinha um tratamento específico.
No depoimento a seguir, sua mãe, a fisioterapeuta Betina Viegas Bender, 43, moradora de São Leopoldo (RS), conta como foi o processo de descoberta da doença, a luta para que governo e sociedade se conscientizem sobre o problema e o alívio que a família sentiu após a menina ser aceita em um teste clínico que resultou na aprovação pela Anvisa de uma terapia enzimática.



"A minha gravidez foi supertranquila, sem nenhuma intercorrência. Quando a Isadora nasceu, tudo parecia perfeitamente bem, mas a partir dos 6 meses de vida, quando começamos a introduzir outros alimentos na dieta, além do leite, ela começou a ter diarreia e vômito e percebemos que seu abdome estava muito globoso.
Procuramos um médico e ele diagnosticou intolerância à lactose. Modificamos a sua alimentação, mas o quadro não melhorava. Pelo contrário, só piorava. Naquela época, ela também passou a ter infecções respiratórias seguidas.
Teve um dia que precisamos correr para o pronto-socorro e a médica que nos atendeu disse que o abdome dela estava alterado, com o baço e o fígado muito grandes, e que seria bom procuramos o nosso pediatra para fazer uma investigação. A partir daí, iniciamos uma pequena saga.
Fomos ao pediatra e ele viu que tinha mesmo alguma coisa errada. A suspeita foi de leucemia e nos indicou um hematologista. O hemato, quando examinou a Isadora, descartou de cara a leucemia e nos sugeriu um gastropediatra.
Conseguimos consulta com uma médica que tinha acabado de fazer especialização em doenças genéticas. Ela disse que tinha cara de ser glicogenose (doença genética e hereditária que afeta o metabolismo do glicogênio) e nos encaminhou para outra especialista, a geneticista Carolina Fischinger Moura de Souza, do HCPA (Hospital de Clínicas de Porto Alegre).
Primeiro, a doutora investigou a suspeita de glicogenose, mas deu negativo. As outras hipóteses foram doença de Gaucher e ASMD. A Isadora precisou fazer alguns exames genéticos e o resultado foi ASMD tipo B, uma patologia genética rara causada por uma deficiência da atividade enzimática e que resulta no acúmulo de uma espécie de gordura nas células.
Do diagnóstico ao tratamento
Recebemos o laudo no dia 12 de março de 2014, mais ou menos seis meses depois que a Isadora começou a apresentar os primeiros sintomas e quando ela já estava com quase 1 ano de idade. No nosso caso até que foi rápido, porque muitas pessoas levam anos para conseguirem um diagnóstico de doença rara.
Mas ainda assim foi um baque, fiquei sem chão. Tudo o que lia sobre este problema era muito ruim, negativo. Foi um período bastante assustador, ainda mais porque não havia tratamento. O que podíamos fazer era apenas tentar controlar algumas das alterações.

Naquela época, a minha filha estava com o fígado e o baço bem aumentados. Ela também apresentava baixa estatura e tinha a dentição atrasada e a musculatura um pouco fraca, flácida, se cansava fácil e estava com triglicerídeos e colesterol muito altos e a imunidade baixa.
Um dia, a doutora Carolina nos disse que um estudo de uma medicação injetável, uma enzima para a ASMD, estava sendo realizado fora do país, mas que ainda não tinha previsão de chegada ao Brasil. Minha irmã foi para Londres na mesma época para uma temporada de três meses. Lá, o estudo estava acontecendo e tentamos incluir a Isadora nele, mas não deu certo.
Nisso, os anos passaram e ela foi se desenvolvendo, mas a sua barriga foi ficando cada vez maior, bem globosa, parecia uma gravidez de 9 meses em uma criança de 3 anos. O fígado e o baço chegaram a atingir o tamanho de um adulto, o que comprometeu a movimentação e o equilíbrio.
O sistema imunológico também não estava funcionando bem, tanto que uma vez fomos para a praia e ela contraiu uma bactéria simples, mas que o seu organismo não conseguiu combater. Foi preciso fazer um tratamento mais intensivo e ela ficou 10 dias internada.
Fizemos tudo o que pudemos naquele momento para chamar a atenção dos governantes e da sociedade para a doença. Só que o desespero bateu, até o dia em que finalmente o estudo da medicação foi aprovado no Brasil, para ser realizado no laboratório do HCPA.
Depois de passar por várias etapas, já que a exigência para participar da pesquisa é bem alta, a Isadora foi incluída e se tornou a primeira criança no mundo a receber a enzima. Foi um alívio e celebramos muito.
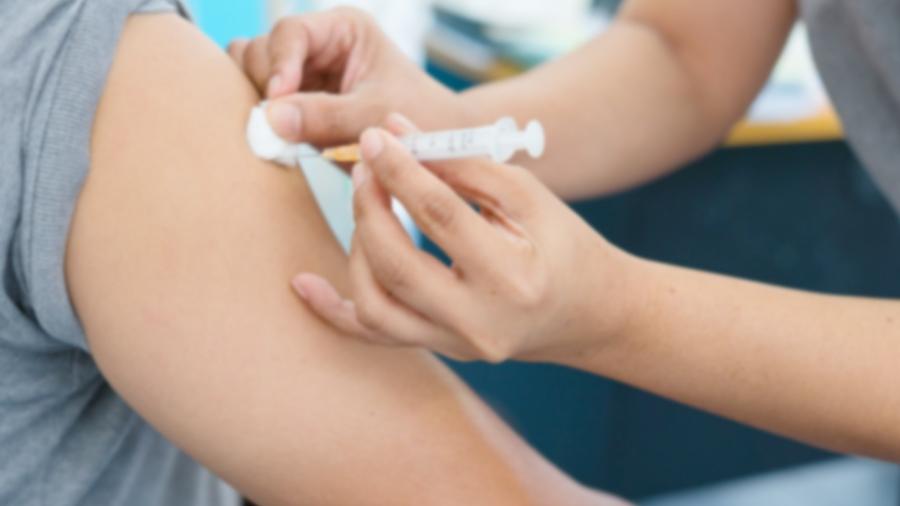
O remédio é administrado de forma injetável e diluído no soro. A cada 15 dias ela passa 4 horas tomando. No primeiro ano do tratamento, já observamos uma mudança enorme. A barriga diminuiu completamente. Hoje, até brincamos que ela tem barriga de tanquinho.
Olhando para ela, você não diz que tem alguma doença. Apesar disso, ainda tem o fígado e o baço um pouco aumentados e o nível de plaquetas segue baixo. Uma pequena parte do pulmão dela também está comprometida.
Esse medicamento que ela toma foi aprovado pela Anvisa em setembro de 2022. A Isadora terá de recebê-lo para sempre —bem como fazer acompanhamento constante— e, pelo fato de ter participado do estudo, o laboratório o disponibilizará gratuitamente e de forma vitalícia.
Dias difíceis

No período em que ficou nos testes clínicos no HCPA, de 2016 até este ano, a Isadora, além de receber a enzima de 15 em 15 dias, tinha de fazer uma bateria de exames a cada 6 meses, que incluía raio-X, ressonância, tomografia, ecocardiograma e exames de sangue.
Nestes momentos precisávamos nos preparar psicologicamente, porque alguns dias eram bem difíceis. Teve vezes, por exemplo, que levamos quase 1 hora tentando convencê-la e dar o braço para achar o acesso venoso. Ela sofre e nós acabamos sofrendo junto, mas sabemos que o benefício é muito maior.
Eu e meu marido sempre procuramos tratar a doença como algo natural. Só que, de uns tempos para cá, como ela está crescendo e se tornando um ser mais independente, começou a questionar por que isso aconteceu com ela.
Explicamos que nem todo mundo nasce igual e que não é uma punição. Também nunca escondemos das pessoas. Pelo contrário, queremos que elas saibam para entender e acharem normal.
Mas claro que já vivemos situações ruins. Lembro de duas viagens para Gramado. Em uma delas, uma senhora olhou para a Isadora e depois nos perguntou: 'Vocês não dão remédio de verme para essa criança?' Na hora ficamos completamente sem reação.
Em outra vez, três pessoas estavam em uma mesa e, quando passamos, uma delas disse: 'Olha, essa aí está com a barriga de chope maior que a minha'. Dessa vez a minha irmã reagiu.
Antes do medicamento, ainda tivemos de lidar com uma vida bastante regrada. Tínhamos que evitar que a Isadora caísse por causa do tamanho do baço e do fígado —eles estavam muito expostos.
Mesmo hoje ela precisa de certos cuidados. E, para que lide melhor com esse monte de coisas, faz terapia desde pequena.
Isso, inclusive, foi muito importante durante a pandemia, porque tivemos de mantê-la em isolamento, só tendo contato com os profissionais que administravam a medicação. Como a imunidade dela é baixa e há o comprometimento pulmonar, não sabíamos o que poderia acontecer se contraísse o vírus da covid-19.
Acesso para todos

Por causa da doença da Isadora, eu e meu marido decidimos não ter outro filho. Como a chance de o bebê nascer com a doença é alta, de 25%, por ser tratar de um problema autossômico recessivo, achamos melhor não arriscar.
Hoje, nossos esforços são focados mesmo na luta para divulgar a ASMD. E, desde que recebemos o diagnóstico, minha família toda se envolveu nesse trabalho.
Atuamos sem parar nos últimos anos para sermos ouvidos, chamarmos a atenção das autoridades e até dos médicos e levarmos mais informações para as pessoas.
Queremos que todos os pacientes consigam ter acesso ao tratamento, assim como a Isadora conseguiu. Antes de ela entrar para o estudo, pelo quadro que apresentava, os sintomas que tinha, acredito que era grande a chance de perdê-la em algum momento.
Esse era o meu medo, não estava preparada para isso. Mas a medicação salvou a vida dela, e vai salvar a de muitas outras pessoas. Só é preciso que todos tenham acesso."
ASMD é doença genética ultrarrara
A ASMD, tradicionalmente conhecida como doença de Niemann-Pick, faz parte de um grupo de patologias genéticas ultrarraras, as de depósito lisossômico, que interferem no funcionamento do lisossomo (organela intracelular que promove uma espécie de reciclagem do material que circula dentro da célula).
Ela ocorre quando há falta ou deficiência na atividade da enzima esfingomielinase ácida. O que acontece é que, sem ela, não ocorre a quebra do lipídio esfingomielina. Ele, então, acaba acumulando em órgãos como fígado, baço e pulmão e, ao longo do tempo, leva a uma série de complicações clínicas, algumas bem graves.
Como explica Carolina Fischinger Moura de Souza, geneticista do Serviço de Genética Médica do HCPA, vice-presidente da SBTEIM (Sociedade Brasileira de Triagem Neonatal Erros Inatos do Metabolismo) e médica responsável pelo acompanhamento de Isadora, a enfermidade é causada por uma mutação genética que provoca um erro no gene SMPD1, responsável pela fabricação da esfingomielinase ácida.

"Esse gene deixa de funcionar e não produz a enzima", relata a especialista. "Nesta doença, que acomete 1 em cada 200 mil recém-nascidos, vemos quadros de manifestação bem ampla, desde alterações no tamanho dos órgãos, passando por funcionamento ruim da medula, resultando em plaquetas baixas e anemia, até retardo de crescimento. É um problema progressivo e que compromete o funcionamento de órgãos e sistemas."
Para se obter o diagnóstico, primeiro é preciso haver uma suspeita. A partir daí, é feita a dosagem da esfingomielinase ácida através de exame de sangue —se ela estiver com uma atividade abaixo de 10% do normal, já se pode caracterizar a patologia. Outro método de investigação é a análise genética do DNA, para verificar a mutação no gene SMPD1.
Crônica e progressiva, a enfermidade não tem cura e até pouco tempo não tinha um tratamento específico, apenas cuidados paliativos. Mas isso mudou em setembro deste ano, quando a Anvisa aprovou o Xenpozyme (olipudase alfa), terapia de reposição enzimática desenvolvida pelo laboratório Sanofi. Ela é feita por meio de uma infusão sanguínea, ou seja, dentro da veia do paciente, e tem como objetivo repor a falta completa ou parcial da enzima.
"Os testes clínicos comprovaram a eficácia da medicação e muitos pacientes foram beneficiados", afirma a vice-presidente da SBTEIM. "Isso é uma vitória não apenas para eles, mas para todos, pois é mais um tratamento que conquistamos para doenças raras, que no geral são tão escassos", finaliza a médica.











ID: {{comments.info.id}}
URL: {{comments.info.url}}
Ocorreu um erro ao carregar os comentários.
Por favor, tente novamente mais tarde.
{{comments.total}} Comentário
{{comments.total}} Comentários
Seja o primeiro a comentar
Essa discussão está encerrada
Não é possivel enviar novos comentários.
Essa área é exclusiva para você, assinante, ler e comentar.
Só assinantes do UOL podem comentar
Ainda não é assinante? Assine já.
Se você já é assinante do UOL, faça seu login.
O autor da mensagem, e não o UOL, é o responsável pelo comentário. Reserve um tempo para ler as Regras de Uso para comentários.